Abstract
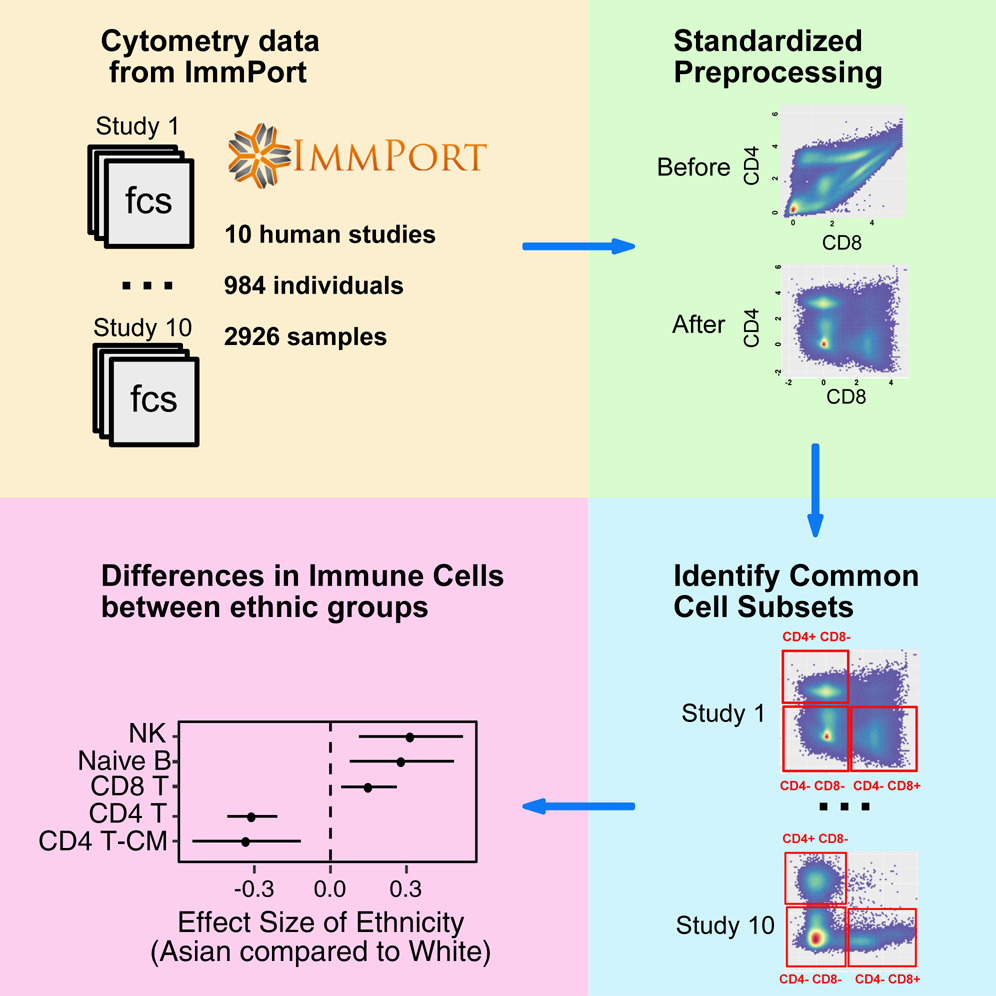
While meta-analysis has demonstrated increased statistical power and more robust estimations in studies, the application of this commonly accepted methodology to cytometry data has been challenging. Different cytometry studies often involve diverse sets of markers. Moreover, the detected values of the same marker are inconsistent between studies due to different experimental designs and cytometer configurations. As a result, the cell subsets identified by existing auto-gating methods cannot be directly compared across studies. We developed MetaCyto for automated meta-analysis of both flow and mass cytometry (CyTOF) data. By combining clustering methods with a silhouette scanning method, MetaCyto is able to identify commonly labeled cell subsets across studies, thus enabling meta-analysis. Applying MetaCyto across a set of ten heterogeneous cytometry studies totaling 2,926 samples enabled us to identify multiple cell populations exhibiting differences in abundance between demographic groups. Software is released to the public through Bioconductor (http://bioconductor.org/packages/release/bioc/html/MetaCyto.html).
Where applicable, full text and supplement provided for fair use.